Компании-разработчики лекарственных средств тратят большие деньги на исследования. Так, в таблице топ-бюджетов R&D все компании раздела «Healthcare» – фармацевтические компании, что занимаются поиском новых лекарств. Дело в том, что поиск лекарственного средства сродни поиску иголки в стоге сена. Ускорить этот поиск и сэкономить помогают информационные технологии. Роль ІТ при поиске новых лекарств частично освещена на Хабре статьями разделов «биоинформатика» и «биотехнологии», в том числе этой . Однако читателям представлен только подход со стороны биологии. Здесь же предлагается более полный обзор с учетом достижений со стороны химии. Если интересно – поехали!
Для начала ответим на вопрос: что нужно чтоб лекарство подействовало? Надо чтоб молекула лекарственного вещества сначала
1) прошла сквозь биологические системы к своей биомишени — например, веществу с таблетки нужно миновать пищеварительный тракт (всасывание), систему кровообращения (распределение), печень (метаболизм) и притом не успеть вывестись почками (элиминация), а потом
2) провзаимодействовала с биомишенью – этой мишенью обычно является соответственный белок.
Поиск всегда начинают со второго пункта – никому не нужно вещество, которое «проходить» организм может, а действовать – нет. Поскольку с одной стороны у нас маленькая молекула химического вещества, а с другой – большая молекула белка, то и подходить можно с двух разных сторон.
Подход со стороны мишени (target-based, «биологический» подход) подразумевает, что мы сначала выбираем себе биомишень, кристаллическая структура которой известна, и потом исследуем возможность ее взаимодействия с множеством молекул, о которых мы можем ничего не знать (то есть даже если есть какая-то информация о их свойствах – она не используется). Сама возможность взаимодействия изучается с помощью так называемого докинга (docking). Если кратко, то процедура следующая: находим структуру интересующего нас белка, кристаллизованного вместе с известным активным веществом (.pdb файл, например, на сайте rcsb.org), открываем подходящей программой (которая способна визуализировать структуру белка), удаляем оттуда лиганд (этим словом называют вещество, которое взаимодействует с большой молекулой), создаем или находим базу нарисованных молекулярных структур (виртуальная библиотека), и проверяем каждую молекулу насколько хорошо она вписывается в освобожденный карман (pocket). То есть ищем ее наиболее выгодное положение в кармане с помощью градиентного спуска (и его усовершенствований) или (реже) генетического алгоритма. Сложности: 1) структуру многих белков сложно получить, поскольку не получается их кристаллизовать; 2) у маленькой молекулы может быть множество вариантов пространственного расположения (конформаций), более того, при взаимодействии с белком конформация может менятся (induced conformation).
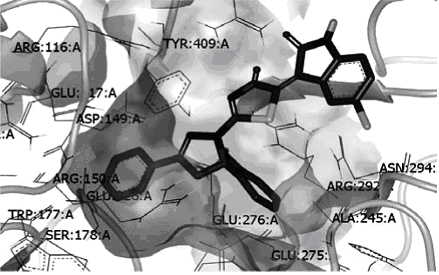
Есть еще один способ – молекулярный дизайн (хотя под этой фразой могут подразумевать многое, именно здесь она наиболее уместна). Вместо того, чтоб совать в карман готовые молекулы, в тех местах где проходило взаимодействие с известным лигандом, оставляют молекулярные фрагменты (те самые или другие, которые способны взаимодействовать таким же способом – их называют биоизостерические). А потом стараются соединить фрагменты, вставляя между ними атомы, да так, чтоб получившаяся конформация была устойчивой. Правда достаточного распространения этот метод не нашел. Сложность, по видимому, в алгоритмическом решении такой задачи.
Подход со стороны лиганда (ligand-based, «химический» подход) подразумевает, что у нас есть информация о активности ряда соединений. И мы хотим найти еще более активные. Что прячется под словом «активность»? Это могут быть как количественные, так и качественные результаты исследований реакции на соединение живых сущностей – in vivo (мышь, изолированный орган или ткань, микроорганизм, культура клеток и т.д.), или неживых – in vitro (молекулы белка). При этом мы можем не знать структуру белка, более того, можем вообще не знать биомишени, имея только отклик организма или клеток на присутствие соединения. Смысл подхода в том, что, используя сведения о структуре активных и неактивных соединений, мы можем интерполировать и экстраполировать (в разумных границах) результаты на новые, еще не синтезированные соединения. Здесь опять есть несколько вариантов: фармакофорное моделирование и моделирование количественной связи структура-активность.
Фармакофор – совокупность молекулярных фрагментов с определенным пространственным размещением, присутствие которых делает молекулу активной. Вместо самых молекулярных фрагментом чаще используются фармакофорные «нотации», то есть абстракции, которые включают в себя фрагмент вместе с возможными биоизостерами (например «донор водородной связи», «акцептор водородной связи», «гидрофобная группа», «ароматический фрагмент»,"π-связь" и т.д.). Для каждой возможной комбинации «нотаций» опять-же по принципу градиентного спуска ищутся лучшие координаты для сфер «нотаций», так чтоб подходящие фрагменты активных соединений попадали в соответствующие сферы, а фрагменты неактивных – не попадали. Сложности: точное определение 3D-геометрии молекул для множества конформаций, необходимость в значительном количестве уже протестированных соединений.

Моделирование количественной связи структура-активность (Quantitative Structure-Activity Relationship, QSAR) – если описать структуры соединений с помощью численных характеристик, задача поиска связи сводится к типичной задачи регрессии (если отклик – активность – непрерывная величина) или классификации (если отклик – дискретная номинальная величина). Эти численные характеристики называют молекулярными дескрипторами, их обычно рассчитывают с помощью готового софта (Dragon, CDK, MOE, Accelrys discovery и др.). При этом существует градация дескрипторов от 0D к 3D в зависимости на каком уровне детализации представляется структура молекулы. Примерами дескрипторов могут быть: 0D – количество атомов углерода, молекулярная масса, 1D- количество гидроксильных групп, 2D – дескрипторы, основанные на теории графов, напр. индекс Винера, собственные величины матриц смежности, 3D – квантовохимические дескрипторы, такие как энергия высшей занятой молекулярной орбитали, теплота образования. Некоторые источники выделяют еще 4D-дескрипторы – это векторы различных потенциалов (электростатического, стерического) рассчитанные в пространственной сетке точек. QSAR моделирование с использованием последних называют 3D-QSAR. Как методы машинного обучения наиболее распространены множественная линейная регрессия, проекция на латентные структуры, нейронные сети (как для регрессии так и для классификации) и Random Forest. Другие алгоритмы таже находят применение, но реже. Поскольку число молекулярных дескрипторов часто намного больше чем исследованных соединений, используются методы выбора переменных – генетический алгоритм, пошаговая регрессия, или отсеивания, например на основе отсутствия корреляции с откликом. Сложности: необходимость в значительном количестве уже протестированных соединений, 3D и 4D дескрипторы зависят от точного определения 3D-геометрии молекул.
Докинг, фармафорные и QSAR модели далее используют для проведения виртуального скрининга (или in silico скрининга). То есть не имея соединения синтезированного в реальности, оценивают его активность, и, таким образом, огромное число заведомо неактивных соединений отсеивается, а соединения с предсказанной высокой активностью синтезируют и направляют на биологические исследования. Если соединение активно и не токсично – далее исследуют его фармакокинетические свойства (всасываемость, объём распределения, скорость биотрансформации и элиминации – комплекс ADME). Для предсказания ADME также можно использовать QSAR-моделирование, но это уже совсем другая история.
Бонус не в тему: а еще в странах СНГ химию лекарственных средств (ту что за границей medicinal chemistry) как перевели ошибочно «медицинская химия» – так и употребляют поныне.
Для начала ответим на вопрос: что нужно чтоб лекарство подействовало? Надо чтоб молекула лекарственного вещества сначала
1) прошла сквозь биологические системы к своей биомишени — например, веществу с таблетки нужно миновать пищеварительный тракт (всасывание), систему кровообращения (распределение), печень (метаболизм) и притом не успеть вывестись почками (элиминация), а потом
2) провзаимодействовала с биомишенью – этой мишенью обычно является соответственный белок.
Поиск всегда начинают со второго пункта – никому не нужно вещество, которое «проходить» организм может, а действовать – нет. Поскольку с одной стороны у нас маленькая молекула химического вещества, а с другой – большая молекула белка, то и подходить можно с двух разных сторон.
Подход со стороны мишени (target-based, «биологический» подход) подразумевает, что мы сначала выбираем себе биомишень, кристаллическая структура которой известна, и потом исследуем возможность ее взаимодействия с множеством молекул, о которых мы можем ничего не знать (то есть даже если есть какая-то информация о их свойствах – она не используется). Сама возможность взаимодействия изучается с помощью так называемого докинга (docking). Если кратко, то процедура следующая: находим структуру интересующего нас белка, кристаллизованного вместе с известным активным веществом (.pdb файл, например, на сайте rcsb.org), открываем подходящей программой (которая способна визуализировать структуру белка), удаляем оттуда лиганд (этим словом называют вещество, которое взаимодействует с большой молекулой), создаем или находим базу нарисованных молекулярных структур (виртуальная библиотека), и проверяем каждую молекулу насколько хорошо она вписывается в освобожденный карман (pocket). То есть ищем ее наиболее выгодное положение в кармане с помощью градиентного спуска (и его усовершенствований) или (реже) генетического алгоритма. Сложности: 1) структуру многих белков сложно получить, поскольку не получается их кристаллизовать; 2) у маленькой молекулы может быть множество вариантов пространственного расположения (конформаций), более того, при взаимодействии с белком конформация может менятся (induced conformation).

Есть еще один способ – молекулярный дизайн (хотя под этой фразой могут подразумевать многое, именно здесь она наиболее уместна). Вместо того, чтоб совать в карман готовые молекулы, в тех местах где проходило взаимодействие с известным лигандом, оставляют молекулярные фрагменты (те самые или другие, которые способны взаимодействовать таким же способом – их называют биоизостерические). А потом стараются соединить фрагменты, вставляя между ними атомы, да так, чтоб получившаяся конформация была устойчивой. Правда достаточного распространения этот метод не нашел. Сложность, по видимому, в алгоритмическом решении такой задачи.
Подход со стороны лиганда (ligand-based, «химический» подход) подразумевает, что у нас есть информация о активности ряда соединений. И мы хотим найти еще более активные. Что прячется под словом «активность»? Это могут быть как количественные, так и качественные результаты исследований реакции на соединение живых сущностей – in vivo (мышь, изолированный орган или ткань, микроорганизм, культура клеток и т.д.), или неживых – in vitro (молекулы белка). При этом мы можем не знать структуру белка, более того, можем вообще не знать биомишени, имея только отклик организма или клеток на присутствие соединения. Смысл подхода в том, что, используя сведения о структуре активных и неактивных соединений, мы можем интерполировать и экстраполировать (в разумных границах) результаты на новые, еще не синтезированные соединения. Здесь опять есть несколько вариантов: фармакофорное моделирование и моделирование количественной связи структура-активность.
Фармакофор – совокупность молекулярных фрагментов с определенным пространственным размещением, присутствие которых делает молекулу активной. Вместо самых молекулярных фрагментом чаще используются фармакофорные «нотации», то есть абстракции, которые включают в себя фрагмент вместе с возможными биоизостерами (например «донор водородной связи», «акцептор водородной связи», «гидрофобная группа», «ароматический фрагмент»,"π-связь" и т.д.). Для каждой возможной комбинации «нотаций» опять-же по принципу градиентного спуска ищутся лучшие координаты для сфер «нотаций», так чтоб подходящие фрагменты активных соединений попадали в соответствующие сферы, а фрагменты неактивных – не попадали. Сложности: точное определение 3D-геометрии молекул для множества конформаций, необходимость в значительном количестве уже протестированных соединений.
Моделирование количественной связи структура-активность (Quantitative Structure-Activity Relationship, QSAR) – если описать структуры соединений с помощью численных характеристик, задача поиска связи сводится к типичной задачи регрессии (если отклик – активность – непрерывная величина) или классификации (если отклик – дискретная номинальная величина). Эти численные характеристики называют молекулярными дескрипторами, их обычно рассчитывают с помощью готового софта (Dragon, CDK, MOE, Accelrys discovery и др.). При этом существует градация дескрипторов от 0D к 3D в зависимости на каком уровне детализации представляется структура молекулы. Примерами дескрипторов могут быть: 0D – количество атомов углерода, молекулярная масса, 1D- количество гидроксильных групп, 2D – дескрипторы, основанные на теории графов, напр. индекс Винера, собственные величины матриц смежности, 3D – квантовохимические дескрипторы, такие как энергия высшей занятой молекулярной орбитали, теплота образования. Некоторые источники выделяют еще 4D-дескрипторы – это векторы различных потенциалов (электростатического, стерического) рассчитанные в пространственной сетке точек. QSAR моделирование с использованием последних называют 3D-QSAR. Как методы машинного обучения наиболее распространены множественная линейная регрессия, проекция на латентные структуры, нейронные сети (как для регрессии так и для классификации) и Random Forest. Другие алгоритмы таже находят применение, но реже. Поскольку число молекулярных дескрипторов часто намного больше чем исследованных соединений, используются методы выбора переменных – генетический алгоритм, пошаговая регрессия, или отсеивания, например на основе отсутствия корреляции с откликом. Сложности: необходимость в значительном количестве уже протестированных соединений, 3D и 4D дескрипторы зависят от точного определения 3D-геометрии молекул.
Докинг, фармафорные и QSAR модели далее используют для проведения виртуального скрининга (или in silico скрининга). То есть не имея соединения синтезированного в реальности, оценивают его активность, и, таким образом, огромное число заведомо неактивных соединений отсеивается, а соединения с предсказанной высокой активностью синтезируют и направляют на биологические исследования. Если соединение активно и не токсично – далее исследуют его фармакокинетические свойства (всасываемость, объём распределения, скорость биотрансформации и элиминации – комплекс ADME). Для предсказания ADME также можно использовать QSAR-моделирование, но это уже совсем другая история.
Бонус не в тему: а еще в странах СНГ химию лекарственных средств (ту что за границей medicinal chemistry) как перевели ошибочно «медицинская химия» – так и употребляют поныне.










